Arbeitsgruppe: "Computergestützte Material-, Device- und Prozesscharakterisierung"
Diese Arbeitsgruppe wird unter der Leitung von Dr. Roman Leitsmann in Kooperation mit dem Geschäftsbereich MATcalc der AQcomputare - Gesellschaft für Materialberechnung mbH unter der Geschäftsführung von Dr. Philipp Plänitz bearbeitet.
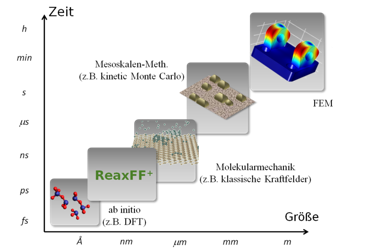
Die rechnergestützte Simulation von Material-, Bauteil- und Prozesseigenschaften nimmt seit Jahren einen festen Platz in fast allen Bereichen der Industrie ein. Eine Reihe von numerischen Lösungsmethoden wie beispielsweise die Methode der finiten Elemente (FEM) im Bereich der Automobilindustrie oder die Hartree-Fock (HF) Methode als Molekular Orbital Theorie im Bereich der Chemie sind bereit altbewährt.Zudem lassen sich heute mittels atomistischer Methoden, wie etwa der Dichte-Funktional-Theorie (DFT), Materialeigenschaften ohne experimentelle Hilfsvariablen von Anfang an (ab initio) berechnen. So lassen sich zudem Parametersätze gewinnen mit denen wiederum automatisiert reaktive Kraftfeldberechnungen Einblick in das physikalisch-chemische Verhalten auf weit größere Skalenbereiche offenbaren. In jeglicher Hinsicht ist ein hinreichendes Verständnis des industriellen Sachverhalts unabdingbar für eine Überführung von Methoden, die bereits im akademischen Umfeld etablierte sind.
Die Arbeitsgruppe setzt den Focus Ihrer Arbeit auf die Anwendung bestehender und die Entwicklung neuer Modelle zur Lösung von skalenübergreifender, multiphysikalischer Problemstellungen aus der Industrie. Durch eine enge Kooperation mit industriellen und experimentellen Forschungsgruppen erfolgt ein kontinuierlicher und zeitnaher Abgleich der Modellierung im Hinblick auf relevante Ergebnisse.
Die rechnergestützte Simulation von Material-, Bauteil- und Prozesseigenschaften nimmt seit Jahren einen festen Platz in fast allen Bereichen der Industrie ein. Eine Reihe von numerischen Lösungsmethoden wie beispielsweise die Methode der finiten Elemente (FEM) im Bereich der Automobilindustrie oder die Hartree-Fock (HF) Methode als Molekular Orbital Theorie im Bereich der Chemie sind bereit altbewährt. Zudem lassen sich heute mittels atomistischer Methoden, wie etwa der Dichte-Funktional-Theorie (DFT), Materialeigenschaften ohne experimentelle Hilfsvariablen von Anfang an (ab initio) berechnen. So lassen sich zudem Parametersätze gewinnen mit denen wiederum automatisiert reaktive Kraftfeldberechnungen Einblick in das physikalisch-chemische Verhalten auf weit größere Skalenbereiche offenbaren. In jeglicher Hinsicht ist ein hinreichendes Verständnis des industriellen Sachverhalts unabdingbar für eine Überführung von Methoden, die bereits im akademischen Umfeld etablierte sind.
Die Arbeitsgruppe setzt den Focus Ihrer Arbeit auf die Anwendung bestehender und die Entwicklung neuer Modelle zur Lösung von skalenübergreifender, multiphysikalischer Problemstellungen aus der Industrie. Durch eine enge Kooperation mit industriellen und experimentellen Forschungsgruppen erfolgt ein kontinuierlicher und zeitnaher Abgleich der Modellierung im Hinblick auf relevante Ergebnisse.
Forschungsthemen
bearbeitet von: Lazarevic, Florian; Schreiber, Michael Prof. Dr.
Im Zuge der voranschreitenden Verkleinerung elektronischer Bauelemente hin zu nanostrukturierten Bauteilen ergeben sich atomskalige Effekte, welche sich nur unter Zuhilfenahme von ab initio Methoden beschreiben lassen. Insbesondere die zuverlässige Vorhersage der elektronischen Eigenschaften eines neuen Materials oder Bauelementes erfordert die Berücksichtigung von quantenmechanischen Effekten.
Anwendungsspektrum:
- Zustandsdichten
- Bandstrukturen
- Charge-Transition-Level (PostDFT Methoden)
- dielektrische Eigenschaften
- Defektbildungsenergien
- Migrationspfade (NEB-Methode)
- Tunnelströme
bearbeitet von: Schreiber, Michael Prof. Dr.
Eine adäquate atomskalige Beschreibung der chemischen und physikalischen Eigenschaften komplexer Prozesse erfordert Zeit- und Längenskalen, welche mit reinen ab inito Methoden nicht erreicht werden können.
Die Simulation solcher Prozesse ist mit einem reaktiven Kraftfeldansatz möglich. Die Beschreibung der inter-atomaren Wechselwirkungen durch empirische, analytische Funktionen ermöglicht die Simulation auf mesoskopischen Zeit- und Längenskalen bei gleichzeitiger exzellenter Genauigkeit. Zur Beschreibung verwenden wir ReaxFF+, eine neu entwickelte reaktive Kraftfeldmethode die auf bindungsordnungsabhängigen Kraftfeldern und einem bindungsordnungsabhängigen Ladungsgleichgewicht basiert. Dadurch wird die korrekte gleichzeitige Beschreibung von rein ionischen und kovalenten Bindungen ermöglicht.Die besondere Herausforderung liegt in der optimalen Wahl der Kraftfeldparameter und der Entwicklung von individuell angepassten materialspezifischen Parametersätzen zur Beschreibung relevanter Fragestellungen.
\r\n Durch die gute Zusammenarbeit mit dem Forschungsdienstleister AQcomputare und dem Software Hersteller QuantumWise werden kontinuierlich problemspezifische Erweiterungen der ReaxFF+ Methode auf Basis chemischer und physikalischer Grundlagen, die im rein akademischen Kontext bisher nicht berücksichtigt wurden, implementiert, verifiziert und validiert.
Anwendungsspektrum:
- Kristallwachstum (MBE, HVPE, CVD, …)
- Reaktionsraten in basischen und sauren Lösungen
- Diffusion komplexer Moleküle
- Phasenübergänge und Kristallisation
- Einfluss von Katalysatoren auf Reaktionsraten
- Erstarren von Polymerschmelzen
- Reibungs- und Abriebprozesse
- Verbrennungsreaktionen in einem Reaktor
- Bruch- und Rissbildung in Verbundmaterialien