Arbeitsgruppen
Bibliometrie
 In der Forschungsdisziplin der Szientometrie wird die Welt der Wissenschaften vermessen. Im Teilgebiet Bibliometrie geht es um die Vermessung von wissenschaftlichen Publikationen, oft mit dem Ziel, Ranglisten von Wissenschaftlern, Instituten, Universitäten oder Ländern aufzustellen. Eine einfache Metrik hierfür ist die Zahl von wissenschaftlichen Publikationen. Diese ist aber nur bedingt aussagekräftig. Die oft vorgenommene Gewichtung mit dem Journal Impact Factor ist aus wissenschaftlicher Sicht unsinnig. In den letzten Jahren wurde es durch die elektronische Verfügbarkeit von umfassenden Zitationsdaten möglich, umfangreiche Zitationsanalysen durchzuführen. Dadurch lässt sich zwar nicht die Qualität der Veröffentlichungen beschreiben, aber zumindest die Sichtbarkeit der Publikationen und damit die Sichtbarkeit eines Wissenschaftlers, Instituts, … In den letzten Jahren ist der 2005 von dem argentinischen Physiker Jorge E. Hirsch vorgeschlagene h-index immer beliebter geworden. In der Professur wird diese Metrik und verschiedene Varianten kritisch untersucht. Bessere Metriken wie z.B. der Vergleich von persönlichen Zitationsraten mit denen einer größeren Gesamtheit werden ebenfalls analysiert.
In der Forschungsdisziplin der Szientometrie wird die Welt der Wissenschaften vermessen. Im Teilgebiet Bibliometrie geht es um die Vermessung von wissenschaftlichen Publikationen, oft mit dem Ziel, Ranglisten von Wissenschaftlern, Instituten, Universitäten oder Ländern aufzustellen. Eine einfache Metrik hierfür ist die Zahl von wissenschaftlichen Publikationen. Diese ist aber nur bedingt aussagekräftig. Die oft vorgenommene Gewichtung mit dem Journal Impact Factor ist aus wissenschaftlicher Sicht unsinnig. In den letzten Jahren wurde es durch die elektronische Verfügbarkeit von umfassenden Zitationsdaten möglich, umfangreiche Zitationsanalysen durchzuführen. Dadurch lässt sich zwar nicht die Qualität der Veröffentlichungen beschreiben, aber zumindest die Sichtbarkeit der Publikationen und damit die Sichtbarkeit eines Wissenschaftlers, Instituts, … In den letzten Jahren ist der 2005 von dem argentinischen Physiker Jorge E. Hirsch vorgeschlagene h-index immer beliebter geworden. In der Professur wird diese Metrik und verschiedene Varianten kritisch untersucht. Bessere Metriken wie z.B. der Vergleich von persönlichen Zitationsraten mit denen einer größeren Gesamtheit werden ebenfalls analysiert.
Computergestützte Material-, Device- und Prozesscharakterisierung
 Diese Arbeitsgruppe wird unter der Leitung von Dr. Roman Leitsmann in Kooperation mit dem Geschäftsbereich MATcalc der AQcomputare - Gesellschaft für Materialberechnung mbH unter der Geschäftsführung von Dr. Philipp Plänitz bearbeitet. Die rechnergestützte Simulation von Material-, Bauteil- und Prozesseigenschaften nimmt seit Jahren einen festen Platz in fast allen Bereichen der Industrie ein. Eine Reihe von numerischen Lösungsmethoden wie beispielsweise die Methode der finiten Elemente (FEM) im Bereich der Automobilindustrie oder die Hartree-Fock (HF) Methode als Molekular Orbital Theorie im Bereich der Chemie sind bereit altbewährt.Zudem lassen sich heute mittels atomistischer Methoden, wie etwa der Dichte-Funktional-Theorie (DFT), Materialeigenschaften ohne experimentelle Hilfsvariablen von Anfang an (ab initio) berechnen. So lassen sich zudem Parametersätze gewinnen mit denen wiederum automatisiert reaktive Kraftfeldberechnungen Einblick in das physikalisch-chemische Verhalten auf weit größere Skalenbereiche offenbaren. In jeglicher Hinsicht ist ein hinreichendes Verständnis des industriellen Sachverhalts unabdingbar für eine Überführung von Methoden, die bereits im akademischen Umfeld etablierte sind.
Die Arbeitsgruppe setzt den Focus Ihrer Arbeit auf die Anwendung bestehender und die Entwicklung neuer Modelle zur Lösung von skalenübergreifender, multiphysikalischer Problemstellungen aus der Industrie. Durch eine enge Kooperation mit industriellen und experimentellen Forschungsgruppen erfolgt ein kontinuierlicher und zeitnaher Abgleich der Modellierung im Hinblick auf relevante Ergebnisse.
Die rechnergestützte Simulation von Material-, Bauteil- und Prozesseigenschaften nimmt seit Jahren einen festen Platz in fast allen Bereichen der Industrie ein. Eine Reihe von numerischen Lösungsmethoden wie beispielsweise die Methode der finiten Elemente (FEM) im Bereich der Automobilindustrie oder die Hartree-Fock (HF) Methode als Molekular Orbital Theorie im Bereich der Chemie sind bereit altbewährt.
Zudem lassen sich heute mittels atomistischer Methoden, wie etwa der Dichte-Funktional-Theorie (DFT), Materialeigenschaften ohne experimentelle Hilfsvariablen von Anfang an (ab initio) berechnen. So lassen sich zudem Parametersätze gewinnen mit denen wiederum automatisiert reaktive Kraftfeldberechnungen Einblick in das physikalisch-chemische Verhalten auf weit größere Skalenbereiche offenbaren.
In jeglicher Hinsicht ist ein hinreichendes Verständnis des industriellen Sachverhalts unabdingbar für eine Überführung von Methoden, die bereits im akademischen Umfeld etablierte sind.
Die Arbeitsgruppe setzt den Focus Ihrer Arbeit auf die Anwendung bestehender und die Entwicklung neuer Modelle zur Lösung von skalenübergreifender, multiphysikalischer Problemstellungen aus der Industrie. Durch eine enge Kooperation mit industriellen und experimentellen Forschungsgruppen erfolgt ein kontinuierlicher und zeitnaher Abgleich der Modellierung im Hinblick auf relevante Ergebnisse.
Diese Arbeitsgruppe wird unter der Leitung von Dr. Roman Leitsmann in Kooperation mit dem Geschäftsbereich MATcalc der AQcomputare - Gesellschaft für Materialberechnung mbH unter der Geschäftsführung von Dr. Philipp Plänitz bearbeitet. Die rechnergestützte Simulation von Material-, Bauteil- und Prozesseigenschaften nimmt seit Jahren einen festen Platz in fast allen Bereichen der Industrie ein. Eine Reihe von numerischen Lösungsmethoden wie beispielsweise die Methode der finiten Elemente (FEM) im Bereich der Automobilindustrie oder die Hartree-Fock (HF) Methode als Molekular Orbital Theorie im Bereich der Chemie sind bereit altbewährt.Zudem lassen sich heute mittels atomistischer Methoden, wie etwa der Dichte-Funktional-Theorie (DFT), Materialeigenschaften ohne experimentelle Hilfsvariablen von Anfang an (ab initio) berechnen. So lassen sich zudem Parametersätze gewinnen mit denen wiederum automatisiert reaktive Kraftfeldberechnungen Einblick in das physikalisch-chemische Verhalten auf weit größere Skalenbereiche offenbaren. In jeglicher Hinsicht ist ein hinreichendes Verständnis des industriellen Sachverhalts unabdingbar für eine Überführung von Methoden, die bereits im akademischen Umfeld etablierte sind.
Die Arbeitsgruppe setzt den Focus Ihrer Arbeit auf die Anwendung bestehender und die Entwicklung neuer Modelle zur Lösung von skalenübergreifender, multiphysikalischer Problemstellungen aus der Industrie. Durch eine enge Kooperation mit industriellen und experimentellen Forschungsgruppen erfolgt ein kontinuierlicher und zeitnaher Abgleich der Modellierung im Hinblick auf relevante Ergebnisse.
Die rechnergestützte Simulation von Material-, Bauteil- und Prozesseigenschaften nimmt seit Jahren einen festen Platz in fast allen Bereichen der Industrie ein. Eine Reihe von numerischen Lösungsmethoden wie beispielsweise die Methode der finiten Elemente (FEM) im Bereich der Automobilindustrie oder die Hartree-Fock (HF) Methode als Molekular Orbital Theorie im Bereich der Chemie sind bereit altbewährt.
Zudem lassen sich heute mittels atomistischer Methoden, wie etwa der Dichte-Funktional-Theorie (DFT), Materialeigenschaften ohne experimentelle Hilfsvariablen von Anfang an (ab initio) berechnen. So lassen sich zudem Parametersätze gewinnen mit denen wiederum automatisiert reaktive Kraftfeldberechnungen Einblick in das physikalisch-chemische Verhalten auf weit größere Skalenbereiche offenbaren.
In jeglicher Hinsicht ist ein hinreichendes Verständnis des industriellen Sachverhalts unabdingbar für eine Überführung von Methoden, die bereits im akademischen Umfeld etablierte sind.
Die Arbeitsgruppe setzt den Focus Ihrer Arbeit auf die Anwendung bestehender und die Entwicklung neuer Modelle zur Lösung von skalenübergreifender, multiphysikalischer Problemstellungen aus der Industrie. Durch eine enge Kooperation mit industriellen und experimentellen Forschungsgruppen erfolgt ein kontinuierlicher und zeitnaher Abgleich der Modellierung im Hinblick auf relevante Ergebnisse.
Kohlenstoffnanoröhrchen
Mechanische, elektronische und optische Eigenschaften von Kohlenstoffnanoröhrchen
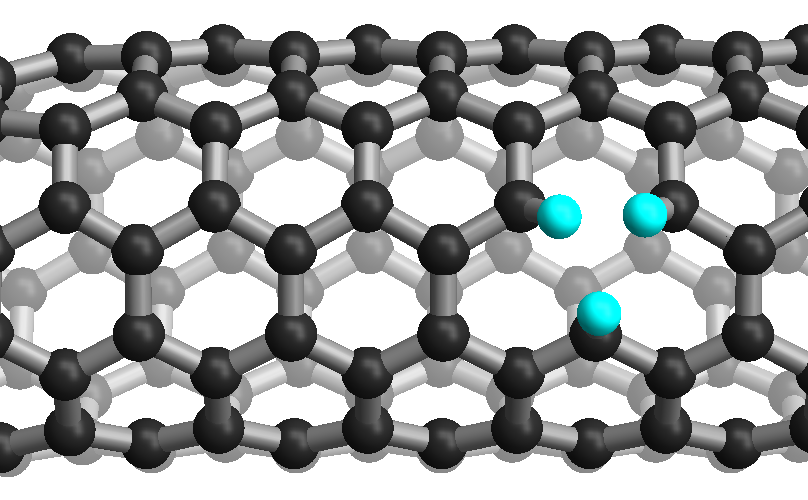 Dieses Forschungsthema wird in Kooperation mit dem Zentrum für Mikrotechnologien (ZfM) und dem Fraunhofer-Institut für Elektronische Nanosysteme (ENAS) in der Gruppe „Simulation von Bauelementen, Prozessen und Anlagen für die Mikro- und Nanoelektronik“ von Dr. Jörg Schuster bearbeitet.
Kohlenstoffnanoröhrchen (engl. carbon nanotubes, CNTs) besitzen hervorragende mechanische und elektronische Eigenschaften, welche zudem durch Variation struktureller Parameter (z.B. Art des CNTs, mechanische Verspannung) manipuliert werden können. Hierin liegt das Potential für zukünftige Anwendungen in den stets kleiner werdenden Mikrochips, z.B. als halbleitender Kanal in Transistoren, als Leitbahn oder als Sensorelement. Ziel der Arbeitsgruppe ist die systematische Beschreibung äußerer Einflüsse auf die mechanischen, elektronischen und optischen Eigenschaften von CNTs – wie z.B. mechanische Verspannung, Defekte oder elektrische Felder. Dabei werden theoretische Methoden verwendet, wie beispielsweise das Tight-Binding-Verfahren (TB), die Dichtefunktionaltheorie (DFT) oder die GW-Methode, die Systemgrößen von 30 bis zu 10000 Atomen erlauben. Darauf basierend können effektive Modelle abgeleitet werden, um den gewünschten Effekt in vereinfachter Weise zu beschreiben, wobei hier auf die Beschreibung mesoskopischer Systeme fokussiert wird. Diese beiden Richtungen bilden eine Basis für Vergleiche mit den Ergebnissen experimenteller Arbeitsgruppen im Physikinstitut und am Zentrum für Mikrotechnologien bzw. Fraunhofer ENAS.
Dieses Forschungsthema wird in Kooperation mit dem Zentrum für Mikrotechnologien (ZfM) und dem Fraunhofer-Institut für Elektronische Nanosysteme (ENAS) in der Gruppe „Simulation von Bauelementen, Prozessen und Anlagen für die Mikro- und Nanoelektronik“ von Dr. Jörg Schuster bearbeitet.
Kohlenstoffnanoröhrchen (engl. carbon nanotubes, CNTs) besitzen hervorragende mechanische und elektronische Eigenschaften, welche zudem durch Variation struktureller Parameter (z.B. Art des CNTs, mechanische Verspannung) manipuliert werden können. Hierin liegt das Potential für zukünftige Anwendungen in den stets kleiner werdenden Mikrochips, z.B. als halbleitender Kanal in Transistoren, als Leitbahn oder als Sensorelement. Ziel der Arbeitsgruppe ist die systematische Beschreibung äußerer Einflüsse auf die mechanischen, elektronischen und optischen Eigenschaften von CNTs – wie z.B. mechanische Verspannung, Defekte oder elektrische Felder. Dabei werden theoretische Methoden verwendet, wie beispielsweise das Tight-Binding-Verfahren (TB), die Dichtefunktionaltheorie (DFT) oder die GW-Methode, die Systemgrößen von 30 bis zu 10000 Atomen erlauben. Darauf basierend können effektive Modelle abgeleitet werden, um den gewünschten Effekt in vereinfachter Weise zu beschreiben, wobei hier auf die Beschreibung mesoskopischer Systeme fokussiert wird. Diese beiden Richtungen bilden eine Basis für Vergleiche mit den Ergebnissen experimenteller Arbeitsgruppen im Physikinstitut und am Zentrum für Mikrotechnologien bzw. Fraunhofer ENAS.
Quasikristalle��������������������������������������������������������������������������������������
Physik aperiodische Legierungen�������������������������������������������������������������������������������������������������������������������������������������������������������������������������
 What is a "quasicrystal"? If one never came across this notion, one might be tempted to guess that this term finds its place among many other "quasi-words" created by modern physics, such as "quasiparticles", "quasimomentum", and so forth. But unlike these, which refer to "virtual" objects whose existence is merely a concept introduced to conveniently describe complex physical systems, a quasicrystal is actually something one can really touch - it is a new state of condensed matter discovered in the early eighties of the twentieth century. The "quasi" in the name refers to the fact that quasicrystals in many respects resemble conventional crystals, but differ from these in one important aspect: They are not built by a single unit cell that repeats periodically in space. It was this lack of periodicity in space, beforehand tacitly assumed for structures with a Bragg-like pure point diffraction, that delayed the acceptance of the experimental discovery and has led to many controversial discussions in the scientific community. Nevertheless, after about fifteen years, the issue appears to be settled today; and quasicrystals have become recognized as solid-state phases, in a way filling a place intermediate between amorphous systems and conventional periodic crystals, which show interesting physical properties.
What is a "quasicrystal"? If one never came across this notion, one might be tempted to guess that this term finds its place among many other "quasi-words" created by modern physics, such as "quasiparticles", "quasimomentum", and so forth. But unlike these, which refer to "virtual" objects whose existence is merely a concept introduced to conveniently describe complex physical systems, a quasicrystal is actually something one can really touch - it is a new state of condensed matter discovered in the early eighties of the twentieth century. The "quasi" in the name refers to the fact that quasicrystals in many respects resemble conventional crystals, but differ from these in one important aspect: They are not built by a single unit cell that repeats periodically in space. It was this lack of periodicity in space, beforehand tacitly assumed for structures with a Bragg-like pure point diffraction, that delayed the acceptance of the experimental discovery and has led to many controversial discussions in the scientific community. Nevertheless, after about fifteen years, the issue appears to be settled today; and quasicrystals have become recognized as solid-state phases, in a way filling a place intermediate between amorphous systems and conventional periodic crystals, which show interesting physical properties.
Ungeordnete Quantensysteme
Metall-Isolator Übergänge, Modelle wechselwirkender und ungeordneter Systeme
 Der Übergang eines Materials vom metallischen in den isolierenden Zustand ist ein bis heute nur unvollständig verstandenes Phänomen. In den letzten Jahrzehnten sind im wesentlichen zwei verschiedene Mechanismen untersucht worden: Zum einen kann ein solcher Metall-Isolator-Übergang (engl: Metal-Insulator-Transition MIT) durch eine Vielteilchenwechselwirkung (Mott-Übergang), zum anderen durch Unordnung (Anderson-Übergang) induziert werden.
Im Anderson Modell bewegen sich unkorrelierte Elektronen auf einem regelmäßigen Gitter, wobei aber die potentiellen Energien an den Gitterplätzen zufällig gewählt werden. Dies führt zu einer Lokalisierung der Einteilchenwellenfunktionen für genügend starke Unordnung und damit zu einem Isolator. Bei schwacher Unordnung sind die Eigenfunktionen räumlich ausgedehnt ähnlich den Blochwellen in einem idealen Kristall. Zur Bestimmung der Phasengrenze zwischen Metall und Isolator im Energie-Unordnungsdiagramm und zur Berechnung der den Übergang charakterisierenden kritischen Exponenten hat sich der Transfermatrix-Algorithmus zusammen mit der Technik des Finite-Size Scaling als nützlich erwiesen.
Der Übergang eines Materials vom metallischen in den isolierenden Zustand ist ein bis heute nur unvollständig verstandenes Phänomen. In den letzten Jahrzehnten sind im wesentlichen zwei verschiedene Mechanismen untersucht worden: Zum einen kann ein solcher Metall-Isolator-Übergang (engl: Metal-Insulator-Transition MIT) durch eine Vielteilchenwechselwirkung (Mott-Übergang), zum anderen durch Unordnung (Anderson-Übergang) induziert werden.
Im Anderson Modell bewegen sich unkorrelierte Elektronen auf einem regelmäßigen Gitter, wobei aber die potentiellen Energien an den Gitterplätzen zufällig gewählt werden. Dies führt zu einer Lokalisierung der Einteilchenwellenfunktionen für genügend starke Unordnung und damit zu einem Isolator. Bei schwacher Unordnung sind die Eigenfunktionen räumlich ausgedehnt ähnlich den Blochwellen in einem idealen Kristall. Zur Bestimmung der Phasengrenze zwischen Metall und Isolator im Energie-Unordnungsdiagramm und zur Berechnung der den Übergang charakterisierenden kritischen Exponenten hat sich der Transfermatrix-Algorithmus zusammen mit der Technik des Finite-Size Scaling als nützlich erwiesen.