The Time-Resoved-Spectroscopy group works on photoinduced dynamical processes
in condensed matter. This processes are investigated by timeresolved optical
spectroscopy and molecular dynamics simulation. At present the following
topics are under study:
1. Energy- and
Charge Transfer in Aggregates of Organic Molecules
The dynamics of energy and charge transfer processes between organic
molecular units in liquid solutions are studied. These molecular units
consist of different kinds of porphyrins and quinones which may be linked
either covalently or by self-aggregation.
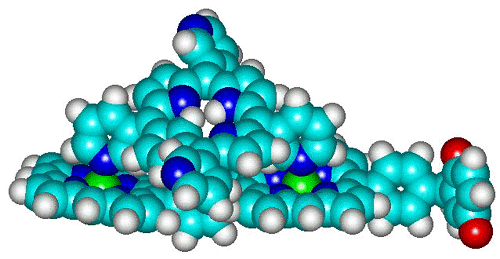
| In a covalently linked dimer consisting of a zinc-porphyrin
(ZnP) and a free-base porphyrin (H2P) energy is transfered from the initially
excited ZnP to H2P on a time scale of a few ps. [91.1]
. |
 |
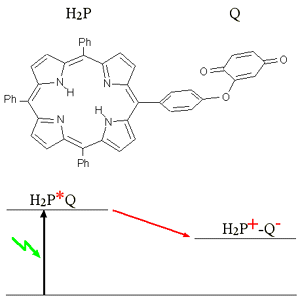
Linking a porphyrin to an
electronacceptor (quinone (Q), pyromilitimid (Pim)) leads to electron
transfer from porphyrin to the acceptor, when the former is optically excited.
In contrast to the case of energy transfer in this case the transfer rate
depends very much on the surrounding medium (dielectric constant of the
solvent, temperature) Performing temperature dependent measurements the
characteristic parameters of these electron transfer processes can be determined.[91.2]. |
|
In a system consisting of two porphyrins and an electron acceptor competition
between energy and charge transfer occurs [90.1].
The most interesting question is whether superexchange processes play a
central role for the charge transfer in these molecular aggregates[95.1]
In order to analyze the influence of the spatial arrangement of the
molecular subunits in these aggregates on the charge transfer processes
a large variety of different supramolecular structures were realized [96.1,
96.2]
and their tranfer properties investigated [92.1,
93.1,
98.1].
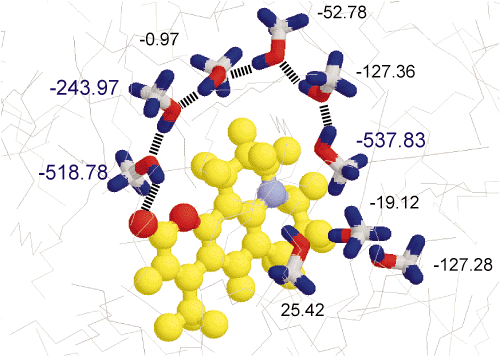
2. Solvation Dynamics
Solvation dynamics are studied in binary mixtures of polar and non-polar solvents. Alcanes are
used as non-polar component while different alcohols or nitriles constitute the polar component. The
dynamics are investigated by monitoring the time-resolved Stokes shift of a suitable probe molecules
dissolved in the solvent mixture. A suitable probe molecule shows a strong change in its static
dipolemoment upon optical excitation and does not exhibit internal dynamics (i.e. photo isomerization).
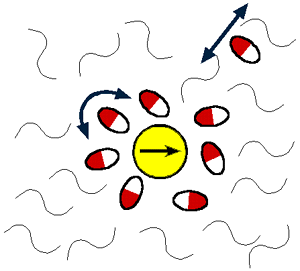
| Up to now we used mostly Coumarin 153 for this purpose. The choice of solvent mixture is made in
order to realize selective solvation of the probe molecule by the polar solvent component. I. e. a
solvent shell of increased concentration (as compared to the bulk) of polar molecules is formed around
the probe molecule. |
|
Selective solvation could be clearly shown for Coumarin 153 in alcane/alcohol mixtures.
Changing systematically the viscosity the non-polar component (length of alcane chain) and the size of
the polar solvent molecules (different kind of alocohol molecules) as well as the concentration of the
polar component the influence of these parameters on the solvation dynamics was studied. It turned out
that on the timescale investigated it is mainly translational diffusion of polar molecules, which
determines the dynamics by increasing the amount of selective solvation in the excited state as compared
to the ground state (dielectric enrichment).[97.1] Thus large Stokes shifts of more than
1000 cm-1 are observed even at very low concentration (less than one vol%) of the polar
solvent. (This Stokes shift is approximately 1/3 of that found in the 100% polar solvent). The
characteristic times (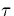 S)
observed for the Stokes shift are strongly dependent on the concentration of
the alcohol and much longer as compared to the pure alcohol. For example it is 1.75 ns in a mixture of
1 mol% of methanol in hexane while it is 5 ps (mean value) in pure methanol. Good agreement is
obtained between the dependence of S
on concentration (c) of the alcohol component
(S ~ c-2/3)
and estimations based on the statistics of diffusion. Further support comes from the
observed linear dependence between S
and the viscosity of the non-polar matrix.
It is not possible to derive any detailed information on the microscopic structure of the solvation
shell around the probe molecule from experimental results. Therefore molecular dynamics simulations have
been carried out. Detailed models of the Coumarin molecule (as opposed to simple spheres often used in
literature) were used for the simulation in order to model specific solute solvent interactions.
The charge distribution and geometry of C153 was calculated using semiempirical quantummechanical
calculations (MOPAC). The simulations were carried out for one molecule of C153, ten molecules of
methanol and 216 molecules of hexane. During equilibrium simulations in the ground (or excited) state
the change in electrostatic energy (
S)
observed for the Stokes shift are strongly dependent on the concentration of
the alcohol and much longer as compared to the pure alcohol. For example it is 1.75 ns in a mixture of
1 mol% of methanol in hexane while it is 5 ps (mean value) in pure methanol. Good agreement is
obtained between the dependence of S
on concentration (c) of the alcohol component
(S ~ c-2/3)
and estimations based on the statistics of diffusion. Further support comes from the
observed linear dependence between S
and the viscosity of the non-polar matrix.
It is not possible to derive any detailed information on the microscopic structure of the solvation
shell around the probe molecule from experimental results. Therefore molecular dynamics simulations have
been carried out. Detailed models of the Coumarin molecule (as opposed to simple spheres often used in
literature) were used for the simulation in order to model specific solute solvent interactions.
The charge distribution and geometry of C153 was calculated using semiempirical quantummechanical
calculations (MOPAC). The simulations were carried out for one molecule of C153, ten molecules of
methanol and 216 molecules of hexane. During equilibrium simulations in the ground (or excited) state
the change in electrostatic energy (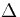 W) upon switching the probe molecule to the excited state (or vice
versa) without changing the solvent surroundings was monitored. This change in electrostatic energy is
responsible for the additional shift and broadening of the static absorption (or fluorescence) spectra.
Simulations were performed for a total of 1.5 ns using times steps of 5 fs. Convoluting the distribution
of W with experimental spectra in pure hexane lead to satisfying agreement with the experimental
spectra in the mixture. [98.4]. Analyzing the trajectories obtained from the simulations shows
pronounced clustering of the methanol molecules. When the C153 is in the ground state they stay apart
from the probe molecule forming clusters of 3 to 4 methanol molecules. With the C153 in the excited
state the methanol molecules form hydrogen bonds to different sites of C153 as well as to each other,
thus forming a strong solvation shell around the probe molecule.
W) upon switching the probe molecule to the excited state (or vice
versa) without changing the solvent surroundings was monitored. This change in electrostatic energy is
responsible for the additional shift and broadening of the static absorption (or fluorescence) spectra.
Simulations were performed for a total of 1.5 ns using times steps of 5 fs. Convoluting the distribution
of W with experimental spectra in pure hexane lead to satisfying agreement with the experimental
spectra in the mixture. [98.4]. Analyzing the trajectories obtained from the simulations shows
pronounced clustering of the methanol molecules. When the C153 is in the ground state they stay apart
from the probe molecule forming clusters of 3 to 4 methanol molecules. With the C153 in the excited
state the methanol molecules form hydrogen bonds to different sites of C153 as well as to each other,
thus forming a strong solvation shell around the probe molecule.